Antibody Tool Catalog
AI-enabled tools for structure prediction, design, docking, simulations, and sequence analysis.
This catalog is generated from your local snapshot and contains 124 tools.
Browse by Category
- Protein Design (44)
- Structure Prediction & Folding (20)
- Sequence Analysis & Annotation (19)
- Ligand/Drug Design & Screening (16)
- Molecular Docking & Interactions (13)
- Utilities & Conversions (11)
- Evolution & Phylogenetics (6)
- Molecular Dynamics & Simulation (4)
- Multi-omics (4)
Tool Directory
-
BioBind
Design antibodies, nanobodies, scFvs, and peptides with high affinity and low immunogenicity.
Supports the design of antibodies, nanobodies, scFvs, and peptides against arbitrary protein or complex targets.
-
BindFilter
Fold and score many binder candidates with a unified, machine-readable output.
Folds each binder candidate independently using the selected folding backend.
-
BioFold2
Design next-generation enzymes by co-optimizing for catalytic activity, stability, and solubility.
Powered by our proprietary BioFold2 model.
-
.webp)
Boltz-2 (AlphaFold3)
Open-source AlphaFold3-class model (Boltz-2) with built-in affinity prediction.
Dedicated affinity-prediction head with near-FEP accuracy for binding-energy estimation.
-
.webp)
Chai-1 (AlphaFold3)
Commercial friendly alternative to AlphaFold3 with competitive MSA-less option.
Delivers AlphaFold3-level accuracy for biomolecular structure prediction.
-
.webp)
Protenix (AlphaFold3)
Another AlphaFold3 implementation developed by the ByteDance team.
Reproduces AlphaFold3's architecture for predicting 3D structures of biomolecular complexes.
-
.webp)
IntelliFold (AlphaFold3)
Open-source AlphaFold3-class model (IntelliFold) with fast, accurate structure prediction across biomolecules.
Custom FlashAttentionPairBias kernel for faster, memory-efficient inference on large biomolecular systems.
-
AlphaFold2
Accurately predict protein and complex structures at the atomic level using their amino acid sequence.
Utilizes the faster & more accurate ColabFold implementation of AlphaFold2.
-
BoltzGen
Generate high-affinity binders for proteins, nucleic acids, and small molecules using an all-atom diffusion model.
Unifies structure prediction and binder design in a single all-atom diffusion framework.
-
BindCraft
One-shot design of functional protein binders.
One-shot design of functional protein binders with high accuracy, powered by deep learning models specialized for protein–protein interactions.
-
.webp)
OpenFold3 (AlphaFold3)
Open-source AlphaFold3 reproduction achieving near-parity accuracy across biomolecular modalities.
Implements AlphaFold3-compatible diffusion-based architecture for multimodal structure prediction.
-
GNINA
Enhanced molecular docking with deep learning.
Integrates CNNs for improved scoring and optimization of ligand binding poses.
-
DynamicBind
Predict protein-ligand complexes using protein structure files and ligands in SMILES format.
Employs a deep equivariant generative model to predict ligand-specific protein-ligand complex structures.
-
DiffDock-L
Dock a ligand onto any protein receptor with high accuracy.
Utilizes the improved DiffDock-L implementation.
-
RFantibody
Generative antibody/nanobody design with fine-tuned RFdiffusion
Epitope-guided de-novo CDR backbone generation and docking via fine-tuned RFdiffusion.
-
RFdiffusion3
All-atom generative diffusion model for designing proteins, nucleic acid binders, and enzymes with precise non-protein interaction conditioning.
Fully atomistic generation creates protein backbones and side chains simultaneously, enabling precise modeling of interactions.
-
RFdiffusion2
Design functional enzymes from their reaction mechanisms using an atom-resolution generative model.
Atom-level active site scaffolding
-

RFdiffusion
Design proteins, binders, and more with this protein diffusion model.
Motif Scaffolding
-
GROMACS Molecular Dynamics
Perform Molecular Dynamics using GROMACS framework, simulating many different solvent solute systems.
Simulates a broad range of molecular systems.
-
gmx_MMPBSA
Calculate binding energetics for GROMACS trajectories using MMPBSA/MMGBSA calculations
Includes MMPBSA and MMGBSA calculations for estimating binding free energy of molecular complexes.
-
ColabDock
Accurately predict protein complexes with specialized restraints.
Dock proteins on proteins using a specialized version of AlphaFold2.
-
RoseTTAFold3
Open-source all-atom foundation model for structure prediction and generative design.
Built on AtomWorks, a modular framework for rapid prototyping and high-quality data processing.
-
RoseTTAFold2
Protein structure prediction that's faster than AlphaFold2 and just as accurate.
Accurately predict protein and multimer / complex structures.
-
RoseTTAFold All-Atom
Protein folding model that supports proteins, nucleotides, ligands, metal ions, and other small molecules.
Predict DNA - protein complexes, protein - protein complexes, and protein - small molecule complexes.
-
GenMol
Generative AI for small molecule design and optimization.
De Novo generation of molecules.
-
PocketFlow
PocketFlow is a Deep Generative Model that generates ligands for target protein binding pockets.
PocketFlow can generate hundreds of small molecules in minutes.
-
EvoEF2
Protein stability and binding energy analysis using EvoEF2.
Protein stability assessment.
-
EvoEF2 Mutant Stability Analysis
Analyze the impact of mutations on protein stability using EvoEF2.
Analyzes the impact of specified mutations on protein stability.
-
Isoelectric Point Calculator
Sequence-only pI/pKa prediction (IPC 1/2).
Classic IPC1 scales (Bjellqvist, EMBOSS, ProMoST, etc.) with optional ALL aggregation.
-
QEPPI
Screen and evaluate early-stage PPI-targeting compounds with a tailored drug-likeness index.
Quantitatively assess drug-likeness specifically for PPI-targeting compounds.
-
ChemBounce
Fragment-based molecular generation and optimization tool.
Iteratively evolves molecules by replacing substructures with bioisosteric fragments.
-
Transcript Assembly
Easily perform transcript quantification using an input fastq file.
Automatically assembles raw reads into valid transcripts without requiring a reference genome.
-
Interleaved FASTQ Splitter
Split interleaved FASTQ into left/right FASTQ with validation.
Splits interleaved paired-end reads into left and right FASTQ outputs.
-
LightDock
Powerful molecular docking algorithm for proteins and nucleotides.
Allows you to dock proteins with peptides, DNA, and other proteins.
-
ESMFold
Accurately predict protein structures at the atomic level using its amino acid sequence.
Includes 3D interactive visualizations of all your folded protein.
-
LigandMPNN
Predict alternative sequences for an input protein structure with high accuracy. Also supports ProteinMPNN and SolubleMPNN.
Sucessor of ProteinMPNN service.
-
ProteinMPNN
Predict alternative sequences for an input protein structure with high accuracy. Also supports SolubleMPNN.
Predecessor to LigandMPNN. Note this service has been largely replaced by LigandMPNN and should no longer be used.
-
NetSolP-1.0
Accurately predict protein solubility and expression / usability from amino acid sequence.
Advanced deep learning: NetSolP utilizes cutting-edge transformers for precise protein solubility and usability prediction.
-
DEAnalysis
Differential Expression Analysis pipeline configured for two-condition experiments.
Volcano Plot of transcripts.
-
ESM-IF1
Predict alternative sequences for an input protein structure with high accuracy.
Allows you to inverse fold any protein or complex of proteins.
-
MIF-ST
Predict alternative sequences for an input protein structure with high accuracy.
Allows you to inverse fold any protein.
-
AfCycDesign
Generates improved cyclic protein structures using a modified AlphaFold network.
Utilizes AlphaFold2 for macrocyclic peptide design.
-
AF2Bind
Accurately predict small-molecule-binding residues using AlphaFold2 pairwise representation.
Predict ligand-binding sites.
-
ScanNet Protein Binding Site Prediction
A geometric deep learning model for predicting binding site probability from a structure.
Predict probabilities of functional sites.
-
AFcluster
Predict conformational substates using AlphaFold2 on multiple sequence alignments.
Predict alternative protein conformations.
-
Prodigy Binding Affinity Prediction
PRODIGY predicts binding affinity and dissociation constants for protein–protein complexes based on their 3D structures.
Accepts 3D structural input of protein–protein complexes in PDB format.
-
PPAP
A structure-aware deep learning model for high-accuracy protein-protein binding affinity prediction (Kd).
Integrates AlphaFold structural insights with ESM sequence representations.
-
SPRINT
SPRINT is a fast, accurate, and scalable deep learning framework for virtual screening of thousands of molecules.
Ultra-fast virtual screening enabling pan-proteome-scale DTI predictions.
-
DNA Chisel Sequence Optimizer
DnaChisel edits DNA sequences to satisfy biological constraints and optimize properties like codon usage, motif distribution, and GC content.
Defines DNA optimization problems using a human-readable, constraint-based DSL.
-
CodonTransformer
A deep learning-based tool for multispecies codon optimization.
Multispecies support spanning 164 organisms across all kingdoms of life.
-
TIsigner Expression Optimization
A nucleotide sequence based method for optimizing protein expression.
Expression optimization through lowering Opening Energy.
-
SoDoPE Solubility Optimization
A sequence based method for optimizing protein solubility.
Fast and accurate optimization of protein solubility.
-
Razor Signal Peptide Detection
A sequence based method for detecting signal peptides.
Signal Peptide detection.
-
WoLF PSORT Protein Localization
Predicts subcellular localization sites from protein sequence.
Uses a modied kNN algorithm to predict the location of protein sequences.
-
ProtNLM
Predict protein annotations from sequence using ProtNLM.
Predict name and function annotations of amino acids sequences.
-
ESM-2 for PTMs
Predict potential post translational modification sites from sequence data.
Use ESM-2 model to find potential PTM sites.
-
EnzBert E.C. Prediction
Enzbert predicts enzymatic classes of protein sequences in batch or individually.
Predict monofunctional enzyme classes from sequence alone.
-
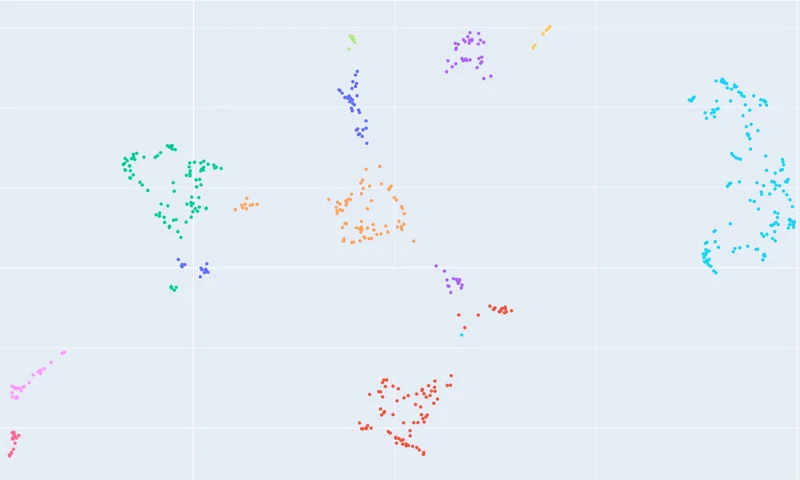
ClusterProt
Cluster same length proteins using only their structures.
Creates a 2D projection and cluster of a set of input proteins of same length.
-

ITsFlexible
A deep learning tool that predicts the conformational flexibility of antibody and T cell receptor (TCR) CDR3 loops, classifying them as 'rigid' or 'flexible'.
Powered by a graph neural network (GNN) architecture.
-
CatPred
A deep learning framework for predicting enzyme kinetic parameters (kcat, Km, Ki).
Predicts turnover numbers (kcat), Michaelis constants (Km), and inhibition constants (Ki).
-
DLKcat Kcat Prediction
Predict Kcats of complexes using a protein sequence and compounds in SMILES format.
Predicts Kcats between a target protein and a selection of compounds.
-
TemStaPro Protein Thermostability Prediction
TemStaPro predicts protein thermostability from sequence at a range of temperatures.
Predicts protein thermostability from sequence data at a range of temperatures.
-
eTox Drug Toxicity Prediction
Predict Toxicity and Synthetic Accessibility from SMILES text or file inputs.
Predicts toxicity of low molecular weight organic compounds from 0 to 1 where 0 is low toxicity probability and 1 is high.
-
ToxinPred Peptide Toxicity Prediction
Predict peptide toxicity from single protein sequences or in batch using an accelerated algorithm.
Uses a unique combination of machine learning methods and motif based approaches to predict peptide toxicity.
-
AlphaFlow
Use AlphaFlow to generate protein structures that closely reflect experimental and physiological conditions.
Generate many protein conformations resembling experimental and physiological ensembles.
-
AMBER Relaxation
Relax a protein structure using an AMBER settling protocol.
Uses OpenMM for molecular dynamics-based energy minimization
-
CryoAtom Cryo-EM Model Builder
CryoAtom builds atomic models from cryo-EM maps using local attention and 3D rotary position embedding, improving model completeness and speed while lowering resolution requirements.
Leverages a local attention mechanism and 3D rotary position embedding for improved accuracy.
-
DeepEMhancer
DeepEMhancer is a deep learning approach for automatic post-processing of cryo-EM maps, performing masking and sharpening in a single step to improve interpretability.
Performs automatic post-processing of cryo-EM maps using a deep learning approach.
-
Aggrescan3D
Structure-based aggregation profiling with Aggrescan3D
Projects aggregation scores onto protein structures using the Aggrescan scale, analyzing residues in user-provided 3D models.
-
DeepViscosity
Predict high-concentration monoclonal antibody viscosity classes.
Predicts viscosity class (Low <= 20 cP, High > 20 cP) for monoclonal antibodies.
-
DockQ
Assess the quality of protein-protein docking models using the native and predicted structure.
Quantitavely assess predicted protein-protein docking models.
-
Protein Fold Stability Prediction
Predict protein stability from structure using ESM-IF.
Predict absolute protein stability using the ESM generative model for protein structures.
-
Foldseek Structural Clustering
Use the Foldseek easy-cluster algorthim to cluster structures using a representative structure.
Structural clustering of PDBs or mmCIFs by creating a structural alignment.
-
Foldtree
Construct phylogenetic trees from protein structures using Foldseek.
Generate phloygenetic trees using a structure-based approach.
-
EpHod Optimal Enzyme pH Prediction
EpHod is a semi-supervised language model that predicts optimal pH for enzymes from sequence alone.
Predict optimal functioning pH for given enzymes from protein sequences.
-
Immune Builder
Design Antibodies, Nanobodies, and T-Cell Receptors using Immune Builder's state-of-the-art generative models.
Design Antibodies, Nanobodies, and T-Cell Receptors from sequences.
-
ANARCI
ANARCI provides standardized numbering and chain classification for antibody and TCR domains.
Standardized numbering of antibody and TCR sequences (IMGT, Kabat, Chothia, Martin, Wolfguy, Aho).
-
ANARCII
ANARCII is a language model–based tool for scalable, accurate numbering and classification of antibody and TCR repertoires.
Language model–driven annotation and classification for immune receptors.
-
PDB Animator
Render animated GIF and MP4 files from multi-model PDB structures.
Automatically extracts every MODEL record from the uploaded PDB structure and visualizes atomic coordinates.
-
Pangolin RNA Splicing Prediction
Pangolin is a deep learning model to predict splice site strength and the impact of genetic variants on RNA splicing in multiple tissues.
Predicts splice site probability and usage for four organs: heart, liver, brain, and testis.
-
DiffAb Antibody Design
Design Antibodies for a target Antigen using the Antigen structure. DiffAb leverages a probabalistic diffusion model.
Design Antigen-specific Antibodies from Antigen structures.
-
PDBFixer
Fix common issues with PDB files such as missing atoms.
Hydrogen Addition: Automatically adds missing hydrogen atoms to protein structures, essential for molecular simulations.
-
PDB-mmCIF Converter
Converts PDB files to CIF / mmCIF files and vice versa
Convert .cif files to .pdb files in the PDB format.
-
PDB-SDF Converter
Converts PDB files to SDF files and vice versa
Convert .sdf files to .pdb files in the PDB format.
-
PDB2PQR
PDB2PQR converts PDB files to PQR format, adding missing atoms and assigning charges for electrostatics calculations.
Converts PDB files to PQR format.
-
AutoDock Vina (smina)
An enhanced fork of AutoDock Vina offering customizable scoring functions, improved sampling, and better performance for molecular docking simulations.
Allows the use of different or user-defined scoring functions for more tailored docking studies.
-
Conformer Generator
Generate conformers for small molecules and ligands using RDkit.
First generates a set number of 3D conformers for the input molecule using the ETKDGv3 method, which is known for producing chemically plausible conformations.
-
mmseqs2 MSA Generation
Rapidly generate diverse and quality MSAs with support for various pairing modes.
Generates high-quality multiple sequence alignments for protein sequences.
-
USalign Structural Alignment
Efficiently produce accurate 3D structural alignments across diverse macromolecular forms and configurations
Performs 3D structure alignments for monomeric and complex structures of proteins, RNAs, and DNAs.
-
LDDT Structural Comparison
Evaluate protein structure quality with a superposition-free local distance difference score
Provides a superposition-free comparison of protein models and reference structures.
-
StaB-ddG
Fast & accurate deep learning model for predicting binding ∆∆G using folding energy principles and a ProteinMPNN-based inverse folding framework.
Thermodynamic parameterization defines binding ∆∆G as a difference in folding free energies.
-
PAMmla
Machine learning models to predict SpCas9 PAM preference from amino acid sequence.
Predicts SpCas9 PAM preference based on amino acid sequence.
-
PAMmla Evolve
Evolve SpCas9 PAM preference using evolutionary algorithms.
Optimizes SpCas9 PAM preference using evolutionary algorithms.
-
AntiFold
Predict alternative sequences for an input Antibodies, Nanobodies, and Antigen-Antibody structures with high accuracy.
Allows you to inverse fold Antibodies, Nanobodies, and Antigen-Antibody complexes.
-
CryoSAMU
CryoSAMU enhances intermediate-resolution cryo-EM maps using a structure-aware multimodal U-Net, integrating map features with protein language model embeddings for faster, high-quality results.
First multimodal network integrating structural information into a 3D U-Net for cryo-EM map enhancement.
-
MUSCLE v5 MSA Generation
Rapidly generate high-quality multiple sequence alignments for protein sequences.
Generates high-quality multiple sequence alignments for biological sequences, with demonstrated accuracy improvements over popular tools like Clustal-Omega and MAFFT.
-
MAFFT MSA Generation
Rapidly generate multiple sequence alignments for protein sequences.
Optimized for fast, accurate alignment of extensive biological sequence datasets.
-
ProteinMPNN-ddG
An unsupervised deep learning model for rapid and accurate prediction of protein stability changes upon mutation, based on an improved ProteinMPNN methodology.
Predicts the effects of point mutations on protein stability (ΔΔG) using an unsupervised method.
-
OmegaFold
Accurate de novo protein structure prediction without reliance on MSAs.
Predicts high-resolution protein structures directly from primary sequences without the need for multiple sequence alignments.
-
FastTree
Rapidly infer maximum-likelihood phylogenetic trees for large sequence datasets.
Uses heuristic methods to accelerate tree construction for large datasets.
-
Mordred Molecular Descriptor Calculator
High-throughput descriptor engine for ML-ready molecular fingerprints.
1800+ 2D and 3D descriptors including topology, electrostatics, and surface area.
-
ADMET-AI
Predict ADMET properties swiftly and accurately using machine learning.
Employs Chemprop-RDKit, a graph neural network architecture, for precise ADMET property predictions.
-
AllMetal3D
Identify likely metal and water binding sites from a protein structure.
Predicts metal-binding and water-binding sites directly from an input protein structure using 3D CNNs.
-
Free Wilson Analysis
Estimate R-group contributions and predict activity across a congeneric series.
Decomposes molecules into R-groups based on a shared core substructure.
-
ThermoMPNN
ThermoMPNN Predicts protein stability changes with precision and efficiency for mutation analysis and design.
Predicts protein stability changes due to single or double point mutations.
-
CAR-Toner
CAR-Toner is an AI tool for rapid prediction of CAR-T tonic signaling by calculating Positively Charged Patch (PCP) scores.
AI-driven prediction of Positively Charged Patch (PCP) scores to quantify CAR tonic signaling.
-
RhoDesign RNA Inverse Folding
Generates RNA sequences with precise structural fidelity and functional diversity for targeted applications
Generates RNA sequences matching specified 3D structural backbones.
-
StrucTFactor
StrucTFactor leverages 3D protein structures for precise transcription factor prediction, outperforming existing methods.
Predicts transcription factors using 3D secondary structural features of proteins.
-
SaProt
SaProt integrates sequence and structure information through a structure-aware vocabulary to predict protein properties accurately.
Combines sequence and structural information into a unified structure-aware vocabulary for enhanced protein analysis.
-
ProGen2
Create protein variants using nothing but the amino acid sequence.
Allows you to generate novel proteins or extend existing proteins.
-
FlowDock
FlowDock predicts protein-ligand structures and binding affinities using geometric flow matching, enabling multi-ligand docking and fast virtual drug screening.
Integrates geometric flow matching to predict protein-ligand structures and binding affinities.
-
ImaPEp Antibody-Antigen Binding Prediction
ImaPEp predicts binding probabilities for antibody–antigen pairs by representing their binding interfaces as 2D images and leveraging convolutional neural networks.
Predicts paratope–epitope binding pairs with high accuracy, achieving a balanced accuracy (BAC) of 0.84 and AUROC of 0.94.
-
Efficient Evolution
A protein language model-based tool for efficient, task-agnostic design of high-functionality protein variants.
Offers a general-purpose approach applicable across diverse protein families.
-
Humatch
A CNN-based tool for rapid, gene-specific humanization and classification of antibody heavy and light chains.
Jointly humanizes antibody heavy (VH) and light (VL) chains for better pairing and stability.
-
DeepImmuno Immunogenicity Prediction
DeepImmuno is a CNN-based model for peptide immunogenicity prediction with state-of-the-art accuracy across viral and cancer datasets.
Beta-binomial scoring layer outputs continuous immunogenicity scores with calibrated confidence.
-
BioPhi
AI-driven antibody humanization + humanness scoring from natural repertoire data.
Accepts antibodies (VH:VL), nanobodies (single domain), and scFvs (VH:VL domains) as input.
-
DR-BERT
Efficiently annotate disordered protein regions with a compact language model.
Pretrained on extensive unannotated protein datasets for enhanced contextual learning.
-
ProSST Mutation Effect Prediction
ProSST predicts protein mutation effects and functions by integrating sequence and structural data via quantized tokens and disentangled attention.
Combines protein sequences and 3D structures via a Transformer with disentangled attention.
-
ParaSurf
ParaSurf is a deep learning framework that predicts paratope binding sites by analyzing molecular surfaces of antibodies/nanobodies to identify antigen-binding regions across the entire Fab/Fv domain.
Predicts antibody-binding sites across the entire Fab region, including CDR loops and framework residues, using balanced sampling to mitigate class imbalance.
-
Kluster
Protein structure clustering and visualization tool using TM-align/US-align structural alignment and dimensionality reduction techniques (UMAP, t-SNE, PCA).
Supports multiple structural comparison metrics (TM-score, RMSD) with both TMalign and USalign alignment tools.
-
AlphaFind
Fast structure similarity search across AlphaFold DB.
Trained from scratch by the Antibody Platform team and does not make any external API requests.
-
Boltz-1 (AlphaFold3)
An open-source version of AlphaFold3 developed by an MIT lab.
Achieves AlphaFold3-level accuracy for predicting 3D biomolecular complex structures.
-
ABACUS-R Sequence Design
Use ABACUS-R to design protein sequences for a given backbone structure using an encoder-decoder model.
Uses an encoder-decoder to predict sequences using local 3-dimensional structural environments.
-
BioFold
Optimize enzyme thermostability, pH stability, solubility, and reaction rate with high accuracy.
Utilizes our proprietary BioFold model to optimize enzymes.