AlphaFold2
Accurately predict protein and complex structures at the atomic level using their amino acid sequence.
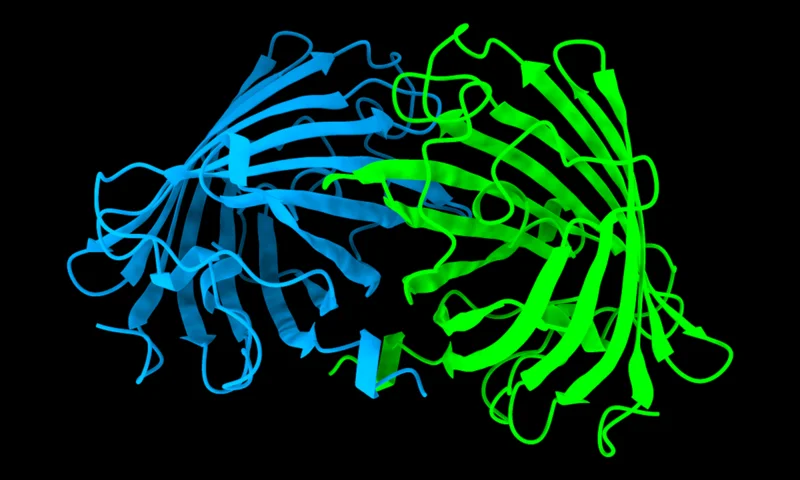
Highly accurate protein structure prediction model that takes an amino acid sequence, MSA, and a template structure as inputs. Both the MSA and template are optional and can be automatically generated. This implementation uses the ColabFold model.
Estimated resource cost: 0.0024025
Categories: Structure Prediction & Folding
Tags: Protein Folding Proteins Protein–Protein Docking
Key Capabilities
- Utilizes the faster & more accurate ColabFold implementation of AlphaFold2.
- Includes 3D interactive visualizations of all your folded protein.
- Includes interactive visualizations for pLDDT and PAE metrics as well as downloads.
- Everything from the protein structure, to the MSA used are available for download.
- Supports monomers and complexes.
- Supports the faster mmseqs2 MSA algorithm.
- Supports different MSA databases, single_sequence, and custom MSA.
- Supports template detection, no templates, as well as custom templates.
- Supports Amber structure relaxation / refinement.
- Supports different pairing modes (unpaired+paired, paired, unpaired).
Runtime Statistics
| Metric | Value |
|---|---|
| runtime_mean | 1392 |
| runtime_median | 315 |
| runtime_std | 6285 |
| runtime_90th_percentile | 2328 |
| runtime_max | 368865 |
Similar Tools
- Boltz-1 (AlphaFold3)
- Boltz-2 (AlphaFold3)
- Chai-1 (AlphaFold3)
- IntelliFold (AlphaFold3)
Ready to submit your job?
Review your configuration, then confirm the estimated credit cost before you run the job. Note that credit estimates are not guaranteed and runtime can vary depending on inputs and settings.
Estimated Credits: 0.0024025
Invite-only, limited-time access. Please contact ztang@getantibody.com.
Invite-only, limited-time access. Please contact ztang@getantibody.com.