Interleaved FASTQ Splitter
Split interleaved FASTQ into left/right FASTQ with validation.
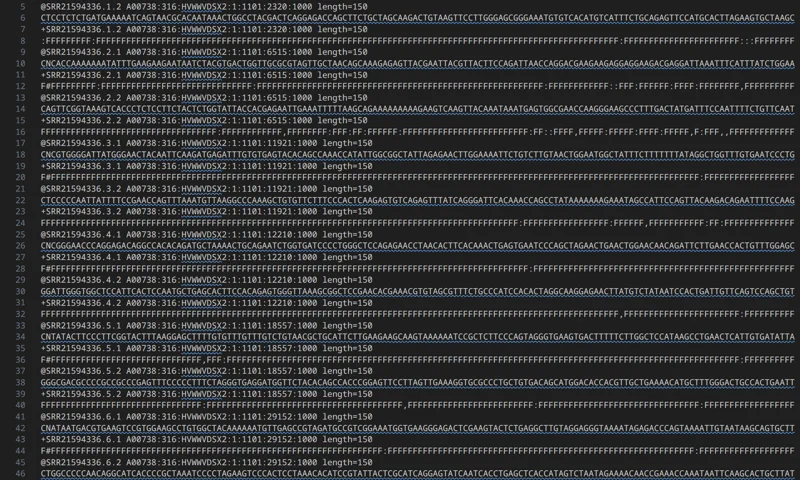
Split an interleaved FASTQ into left/right FASTQ files with strict FASTQ validation. Supports gzip inputs by filename and can preserve original identifiers or rewrite them as @/1 and @/2.
Estimated resource cost: 0.000062
Categories: Multi-omics, Utilities & Conversions
Tags: File Conversion RNA-seq Transcript Assembly
Key Capabilities
- Splits interleaved paired-end reads into left and right FASTQ outputs.
- Validates FASTQ structure and sequence/quality length consistency.
- Supports gzip-compressed inputs via .fastq.gz or .fq.gz filenames.
- Optionally preserves read identifiers or rewrites them as @/1 and @/2.
- Streams input for large files without loading entire datasets into memory.
Runtime Statistics
| Metric | Value |
|---|---|
| runtime_mean | 0 |
| runtime_median | 0 |
| runtime_std | 0 |
| runtime_90th_percentile | 0 |
| runtime_max | 0 |
Similar Tools
- Transcript Assembly
- DEAnalysis
- PDB-SDF Converter
- PDB-mmCIF Converter
Ready to submit your job?
Review your configuration, then confirm the estimated credit cost before you run the job. Note that credit estimates are not guaranteed and runtime can vary depending on inputs and settings.
Estimated Credits: 0.000062
Invite-only, limited-time access. Please contact ztang@getantibody.com.
Invite-only, limited-time access. Please contact ztang@getantibody.com.