gmx_MMPBSA
Calculate binding energetics for GROMACS trajectories using MMPBSA/MMGBSA calculations
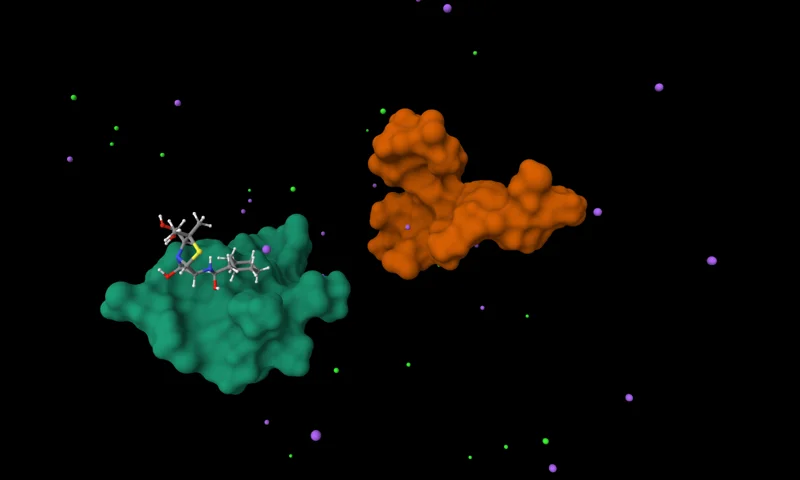
Perform fast MMPBSA/MMGBSA binding free energy analysis on Antibody Platform GROMACS trajectories. Upload files from your prior MD jobs; the service auto-selects decorrelated frames from RMSD, builds the needed index files, and runs Poisson–Boltzmann or Generalized Born calculations for protein–ligand and protein–protein complexes. Outputs include per-frame energy CSVs and concise JSON summaries for each ligand chain or small-molecule pairing.
Estimated resource cost: 0.0005786150000000001
Categories: Molecular Dynamics & Simulation
Tags: Affinity Prediction Energy Minimization Free Energy / Binding Energy Molecular Dynamics
Key Capabilities
- Includes MMPBSA and MMGBSA calculations for estimating binding free energy of molecular complexes.
Runtime Statistics
| Metric | Value |
|---|---|
| runtime_mean | 8011 |
| runtime_median | 4297 |
| runtime_std | 10189 |
| runtime_90th_percentile | 26408 |
| runtime_max | 41985 |
Similar Tools
- GROMACS Molecular Dynamics
- AMBER Relaxation
- AlphaFlow
- BindFilter
Ready to submit your job?
Review your configuration, then confirm the estimated credit cost before you run the job. Note that credit estimates are not guaranteed and runtime can vary depending on inputs and settings.
Estimated Credits: 0.0005786150000000001
Invite-only, limited-time access. Please contact ztang@getantibody.com.
Invite-only, limited-time access. Please contact ztang@getantibody.com.