GROMACS Molecular Dynamics
Perform Molecular Dynamics using GROMACS framework, simulating many different solvent solute systems.
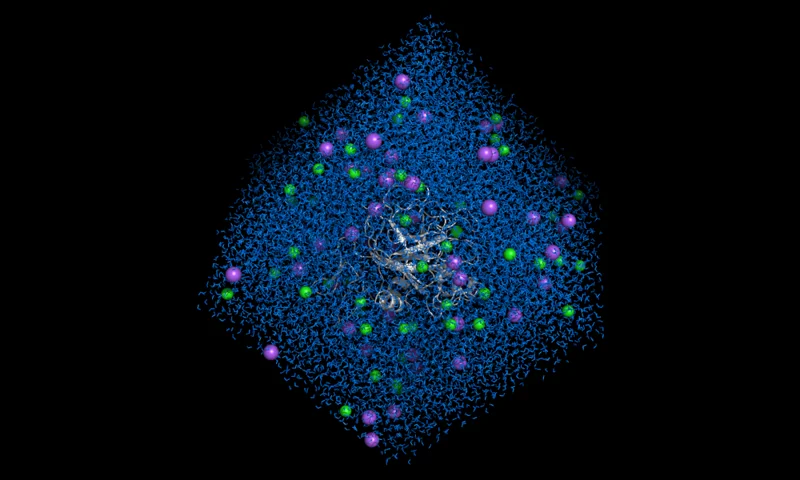
Perform Molecular Dynamics using GROMACS framework, simulating many different solvent solute systems. Simulate proteins and enzymes in different solutions.
Estimated resource cost: 0.0009858
Categories: Molecular Dynamics & Simulation
Tags: Affinity Prediction Energy Minimization Free Energy / Binding Energy Molecular Dynamics
Key Capabilities
- Simulates a broad range of molecular systems.
- Returns many metrics including equilibration steps, RMSD, RMSF, Gyration Radius and more.
- Allows you to specify custom durations for your molecular dynamics simulation in nanoseconds.
- Simulation execution duration may range from hours to overnight. The time to job completion scales with the number of atoms and if the simulation is a protein-ligand interaction.
- Only available for paid users.
Runtime Statistics
| Metric | Value |
|---|---|
| runtime_mean | 16420 |
| runtime_median | 3717 |
| runtime_std | 48348 |
| runtime_90th_percentile | 34511 |
| runtime_max | 739303 |
Similar Tools
- gmx_MMPBSA
- AMBER Relaxation
- AlphaFlow
- BindFilter
Ready to submit your job?
Review your configuration, then confirm the estimated credit cost before you run the job. Note that credit estimates are not guaranteed and runtime can vary depending on inputs and settings.
Estimated Credits: 0.0009858
Invite-only, limited-time access. Please contact ztang@getantibody.com.
Invite-only, limited-time access. Please contact ztang@getantibody.com.